Neuromuscular Notes: New & Emerging Therapies in ALS
06/14/2023
Amyotrophic lateral sclerosis (ALS) is the most frequent adult motor neuron disease. ALS is a phenotypically and biologically heterogeneous disease that affects lower and upper motor neurons, causing progressive weakness and usually leading to death within 2 to 5 years after the onset of symptoms. There are now 4 therapies approved by the Food and Drug Administration (FDA) to slow disease progression or improve survival in ALS. The first 3 medications have a relatively modest effect on survival or disease progression, and more disease-modifying therapies must be developed. The fourth medication, tofersen, was approved very recently for SOD1-mediated ALS. Here, we review the current FDA-approved disease-modifying medications, the challenges in developing new therapies, and selected promising new therapies in clinical trials.
FDA-Approved Medications
The mechanism of action of riluzole, approved in 1995, is highly complex, with multiple effects on neurotransmission and glutamate-induced toxicity to motor neurons. Three clinical trials have shown survival benefit for riluzole and a 2012 Cochrane meta-analysis showed improved survival of 60 to 90 days, but no effect on measures of function or strength.1 Earlier and stable doses of riluzole may provide the largest survival benefit.2 Riluzole is generally well-tolerated, except for mild gastrointestinal upset and occasional elevation of liver enzymes (Table 1).
Edaravone, a scavenger of free radicals, was approved in intravenous form in 2017 and then in oral formulation in 2022. The evidence supporting the use of edaravone in ALS is debated. Edaravone failed to show any benefit in 2 clinical trials. A post hoc analysis of the second trial and a subsequent phase 3 trial have shown that a subgroup of patients with early disease may benefit from edaravone, with a 33% slowing in disease progression at 6 months.3 However, a recent propensity score–matched, nonrandomized clinical trial on long-term treatment with edaravone has failed to demonstrate any benefit at any time, even in the subgroup of patients with early disease.4 Overall, considering the recent availability of oral edaravone and its good tolerance, many physicians and patients consider that even a small benefit may outweigh the risk. Allergic reaction can occur in about 5% of people with asthma and other side effects are rare.
Sodium phenylbutyrate–taurursodiol (PB-TURSO), targeting endoplasmic reticular stress and mitochondria, was approved in 2022 after a phase 2 trial demonstrated both slowing of progression in the Amyotrophic Lateral Sclerosis Functional Rating Scale-revised (ALSFRS-R) and improved survival.5,6 A phase 3 clinical trial is ongoing, and results are expected in 2024. Side effects of the medication include gastrointestinal upset in about 30% of people. Caution should be taken in people with kidney disease or heart failure because of the risk of fluid retention.
The cost of edaravone and PB-TURSO and their overall mild effect on the disease pose issues for patients, health care providers, and insurance coverage. The current recommendations, based on expert opinion, are to propose all 3 medications to all people with ALS. Although recent trials on PB-TURSO allowed for riluzole or edaravone use, data are lacking on the advantages of adding one medication to another. Also, there is no means to predict response to medication or to determine whether people should continue medication indefinitely or if it should be stopped after a short trial. More work must be done to clarify the standard of care for the 3 approved ALS disease-modifying medications.
Challenges in Development of New ALS Therapies
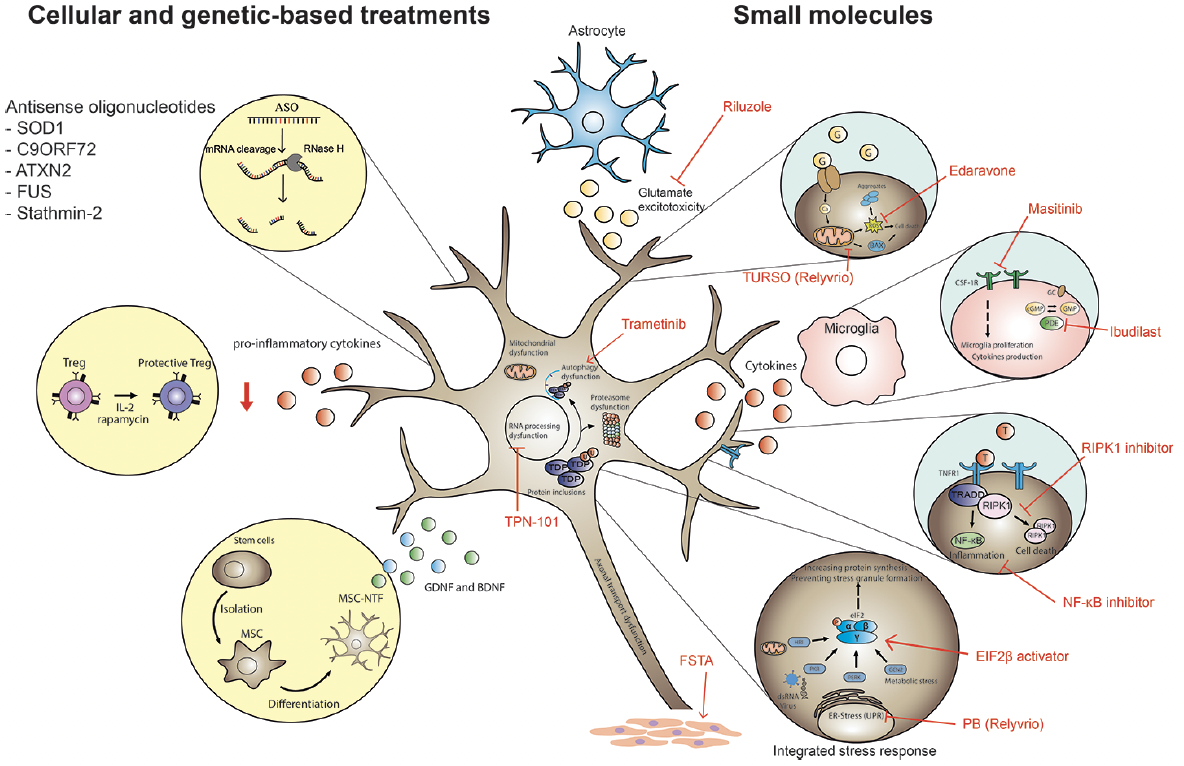
Therapeutic development in ALS faces many challenges, in part because clinical trial design in ALS has been complex because of the genetic, biological, and clinical heterogeneity seen in the ALS population. Many of the potential therapies developed in animal models have failed in clinical trials over the years, but great strides have been made in identifying underlying mechanisms that drive disease pathogenesis, leading to several new therapeutic targets (Figure 1). The large variability in clinical presentation and progression and past exposure to disease-modifying treatments may affect the outcome, presents challenges in the statistical detection of clinical efficacy, and must be considered when designing intervention trials.
ALSFRS-R score is used as the primary outcome in most trials. However, the ALSFRS-R may not be sensitive to clinically meaningful changes in function for all people with ALS. Therefore, more sensitive and objective outcome measures, biomarkers, and prognostic modeling have been identified and used in recent clinical trials. Examples of biomarkers currently used in trials or in development include plasma neurofilament light chain (NfL), cerebrospinal fluid (CSF) SOD1 levels, urinary p75 levels, imaging by PETor MRI scans, and certain electrophysiologic techniques. There are ongoing efforts examining digital biomarkers that can be collected from smartphones or wearable devices to capture changes in real-world behavior and function.
{kind=link}
Figure 1. Identification of underlying mechanisms that drive disease pathogenesis is leading to new therapeutic targets.
ER, endoplasmic reticulum.
Emerging Therapies and Their Targets
Antisense Oligonucleotides
Antisense oligonucleotides (ASOs) are single-stranded DNA that bind to target RNA with improved technology for high affinity and stability. ASOs have been effective in slowing disease progression in spinal muscular atrophy, a form of motor neuron disease. In ALS, ASOs in development work through a mechanism of RNase H-dependent cleavage of the target RNA. To date, genes targeted by ASOs in ALS are SOD1, C9orf72, FUS, Ataxin-2, and stathmin-2, with some new targets on the horizon (Table 2). The recent results of the phase 3 tofersen trial (VALOR [An Efficacy, Safety, Tolerability, Pharmacokinetics and Pharmacodynamics Study of BIIB067 in Adults With Inherited Amyotrophic Lateral Sclerosis]; URL: https://www.clinicaltrials.gov; Unique identifier: NCT02623699) have been of particular interest. Intrathecal boluses of ASO were administered to 72 people with SOD1 ALS. The primary outcome for efficacy—ALSFRS-R change from baseline over 28 weeks—was not met. However, there were favorable trends in secondary outcome measures, particularly in the 39 patients recruited in the rapidly progressive group (eg, changes in SOD1 levels in CSF and plasma NfL).7 The open-label extension group continues to gather more long-term trends and safety data. Likewise, a phase 1 trial of BIIB078 targeting C9orf72, the most common ALS gene, was negative, with a trend toward greater clinical decline observed in people treated with BIIB078 (Table 2).
Autologous Mesenchymal Stem Cells
Cell-based therapies that can target multiple pathways and favor neuronal survival have been studied in many preclinical trials. Clinical trials have been conducted on a single dose of autologous bone marrow–derived mesenchymal stem cells. Mesenchymal stem cell–secreting neurotrophic factors (MSC-NTF) were evaluated in a phase 2 trial of 48 participants and the primary safety outcome was met. CSF markers of inflammation decreased after transplantation of mesenchymal stem cells and monocyte chemoattractant protein-1 (MCP-1) levels correlated with improvement in ALSFRS-R slope up to 6 months.8 These results encouraged a multidose clinical trial of MSC-NTF cells. In a phase 3 study, 196 participants received 3 doses of MSC-NTF cells. MSC-NTF was again found to be safe and well-tolerated, but the primary end point of the rate of change of ALSFRS-R per month was not met. However, CSF MCP-1 and NfL levels demonstrated significant improvement in the treated group.9 Further studies on this safe and well-tolerated treatment are under further investigation.
Targeting Neuroinflammation in ALS
A great deal of evidence suggests a role of neuroinflammation in ALS pathogenesis. Multiple compounds targeting neuroinflammation are in development and are reviewed in Table 2. One pathway of interest is receptor interacting serine/threonine kinase 1 (RIPK1) activation and its implication in cell death and inflammation. RIPK1 is activated by tumor necrosis factor receptor 1 (TNFR 1) and is a regulator of nuclear factor kappa B (NF-κB) signaling. In ALS, increased levels and activation of RIPK1 have been found in postmortem spinal cords of people diagnosed with ALS.10 DNL747, a RIPK1 inhibitor, was studied in ALS and Alzheimer disease, but had potential long-term dose-limiting toxicity.11 A similar compound, DNL788, which is expected not to exhibit the same toxicity, is being studied.
Other Emerging Therapies
Another pathway of interest is the integrated stress response. The integrated stress response is a network of multiple cellular functions and kinases reacting to cellular stress to restore normal homeostasis in the cells. In ALS, kinases implicated in the endoplasmic reticulum stress and unfolded protein responses, including protein kinase R–like endoplasmic reticulum kinase (PERK) and eukaryotic initiation factor 2Β (eIF2Β), are of particular interest. Chronic activation of the integrated stress response and inhibition of eIF2Β are thought to contribute to formation of stress granules and TAR DNA-binding protein 43 (TDP-43) pathologic inclusions.12 Therefore, eIF2Β activators are a therapeutic area of interest in ALS and currently in phase 1 and phase 2 trials.
Increasing muscle function and endurance is an interesting target in ALS and other neuromuscular disorders. Fast skeletal muscle troponin activators increase muscle strength by sensitizing sarcomeres to calcium. Tirasemtiv, a first-generation fast skeletal muscle troponin activator, failed to show significant improvements in a phase 3 trial in ALS, in part because of poor tolerance.13 Therefore, reldesemtiv, a second-generation fast skeletal muscle troponin activator with less blood–brain barrier penetrance and off-target effect, was developed. A phase 2 trial of reldesemtiv failed to meet its primary end point (change in slow upright vital capacity at 12 weeks), but a positive trend was seen in all 3 secondary end points (progression rate of slow upright vital capacity, ALSFRS-R score, and muscle strength megascore).14 A phase 3 clinical trial was terminated recently for futility. In addition, levosimendan, a calcium sensitizer that favors muscle contraction by means of its calcium-dependent interaction with troponin C, was tested in a phase 3 trial, without benefit on respiratory function.15
Platform Trial in ALS
The Healey ALS Platform Trial, the first of its kind, is a perpetual adaptive trial in ALS. In this trial, patients are master screened and then assigned a regimen, with a 3-to-1 randomization within each regimen. The placebo can be shared among the groups for analysis and thus the placebo group is smaller than in traditional ALS trials. A 6-month randomized control period leads to a 6-month open-label extension. The new regimens are introduced more quickly with improved efficiency and reduced costs, facilitating faster results in this innovative approach. The first 4 regimens recently were completed, with none of them meeting their primary end point as measured by the ALSFRS-R, but findings in secondary end points have encouraged ongoing study of CMN-Au8 and pridopidine. Regimen E (trehalose) has enrolled fully and is ongoing. Regimen F (ABBV-CLS-7262) and regimen G (DNL343) started enrollment in early 2023 (Table 3).
Conclusion
Although significant advances have been made in ALS therapies over the past 5 years, ALS remains a terminal disease. Emerging therapies are being developed to target specific pathogenic mechanisms identified in ALS, including antisense oligonucleotides, mesenchymal stem cells, kinase inhibitors, and integrated stress response and endoplasmic reticulum stress inhibitors. The development of ALS therapies needs to be integrated with the development of novel end points that are sensitive to change and will enable us to run faster, more patient-friendly trials while developing a deeper understanding of the disease. There is potential for biofluid and digital biomarkers to play an important role in trials while helping establish an evidence-based standard of care for existing disease-modifying therapies.
Dr. Ho and K.M. Burke have disclosures posted at practicalneurology.com.
Dr. Picher-Martel, S. Luppino, and S. Sullivan report no disclosures.
Facebook Comments